Достаточно редкие виды краниостеноза. Включают в себя как патологию черепных швов, лицевого скелета, так другие аномалии органов и систем организма. При этих вариантов краниостеноза может поражаться как один шов, так и большим количеством швов черепа. Характерным признаком синдромальных краниостенозов является поражение коронарного шва, а так же швов основания черепа, в результате чего основными проявлениями являются брахицефалия и недоразвитие средней зоны лицевого скелета (гипоплазия верхней челюсти, скуловых костей, расщелины лица). В основе классификации синдромальных краниостенозов лежат генетические и этиологические факторы, а так же клинические проявления. Для наиболее часто встречающихся краниостенозов при синдроме Аперта, Крузона, Пфайффера характерно наличие мутации в гене FGFR2, в случае синдрома Пфайффера мутация обнаруживается так же в гене FGFR1. Основными клиническими проявлениями синдромальных краниостенозов кроме внешних признаков – повышение внутричерепного давления с развитием атрофии зрительного нерва и впоследствии слепотой; нарушение носового дыхания по причине нарушения развития воздухоносных путей; нарушение слуха по причине дисфункции евстахиевой трубы; нарушения развития грудной клетки, так же различные варианты синдактилий конечностей.
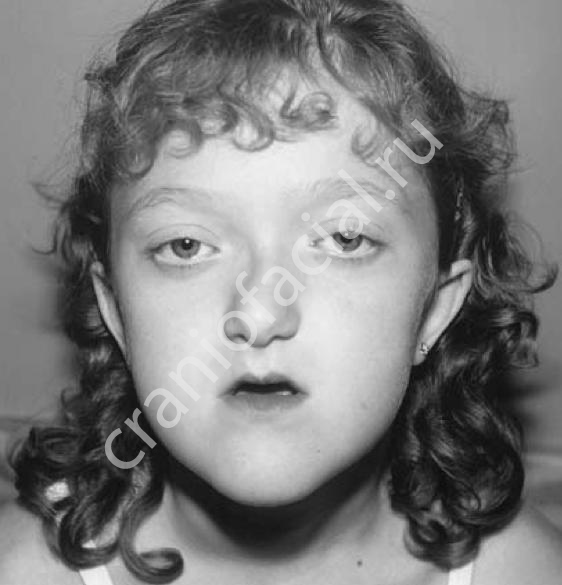
Синдром Крузона или черепно-лицевой дизостоз представляет собой заболевание с аутосомно-доминантным типом наследования. Встречаемость этого синдрома примерно 1 на 25000 новорожденных. В половине случаев причина синдрома – спорадические мутации. Характерна большая вариабельность проявления этого синдрома от лёгких до тяжелых форм. Синдром определяют: наличие синостоза коронарного, а так же сагиттального, лямбдовидного и метопического швов свода и швов основания черепа. Аномалии лицевого скелета включают в себя гипоплазию верхней челюсти, мелкие орбиты, экзофтальм, гипертелоризм, характерна клювовидная форма носа. Часто наблюдается сопутствующее снижение слуха (55%), гидроцефалия, умственная отсталость (последнее время до 3%), эпилепсия (12%), атрофия зрительных нервов, аномалии позвоночного столба (30), крестца и ребёр. У незначительного числа пациентов с синдромом Крузона встречаются анкилозы локтевых суставов и подвывих головки лучевой кости, короткие пальцы.
Синдром Аперта или акроцефалосиндактилия – аутосомно-доминантно наследуемое заболевание. Встречаемость этого синдрома 1 на 160000-200000 новорожденных. Варианты краниостеноза могут быть различными, однако чаще наблюдается вовлечение коронарного шва. Типичны особенности лицевого скелета: гипоплазия средней зоны лица, гипертелоризм, антимонголоидный разрез глаз, косоглазие. Сопутствующие аномалии включают синдактилию, возможны пилоростеноз, эктопия ануса, аплазия легких, различные дефекты сердца (25%), открытый артериальный проток, трахеопищеводный свищ, поликистоз яичника.
Синдром Пфайффера встречается примерно в 1 на 100000 новорожденных. Возникает данный синдром в результате спонтанной мутации в гене FGRFIили II. Ряд авторов считают, что синдром может возникнуть у детей от пожилых отцов. Характерен аутосомно-доминантный тип наследования этого заболевания. То есть достаточно, чтобы один из родителей имел повреждённый ген, чтобы передать его потомкам. Родитель с этим синдромом имеет 50% вероятность рождения ребёнка с такой же патологией. Это характерно и, для описанных выше синдромах Аперта и Крузона. Различают три варианта этого синдрома. Первый (I) тип считается классическим вариантом синдрома, для него характерно наличие краниостеноза – брахицефалии (в большинстве случаев при данном типе синдромального краниостеноза в патологический процесс вовлекаются коронарный, лямбдовидный и сагитталный швы свода черепа) недоразвитие средней зоны лица, аномалия пальцев рук и ног, сохранный интеллект. Более тяжёлыми считаются варианты IIи III, часто дети рождаются нежизнеспособными и погибают вскоре после рождения (в 25-85% случаях), у них выраженный экзофтальм из-за грубого недоразвития средней зоны лица, тяжелая патология конечностей в виде анкилозов локтевых и коленных суставов, аномалий пальцев рук и ног, грубые неврологические нарушения и задержка интеллектуального развития. Отличие между IIи IIIтипами состоит главным образом в варианте краниостеноза и соответственно, форме черепа. При IIтипе череп имеет форму трилистника (клеверный лист, Kleeblattschädel). Существуют и переходные формы, когда нельзя чётко отнести патологию к какому-либо одному из 3-х вариантов. Аномалии рук и ног, характерные для синдрома проявляютя короткими и широкими большими пальцами рук и ног.
Так же дети с синдромом Пфайффера имеют в 50% случаев проблемы со слухом по причине аномально-малого слухового прохода и среднего уха, зрением по причине маленьких глазниц и аномального расположения структур глазницы, а так же по причине повышенного внутричерепного давления, мальформации Киари. Причём частота встречаемости этих нарушений увеличивается при типах от I до III. Ма
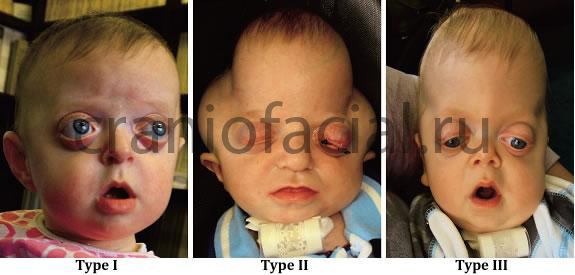
Этот вариант краниостеноза можно диагностировать при внутриутробном УЗИ исследовании, для него характерно наличие экзофтальма, гипертелоризма и широкие большие пальцы рук и ног, определяемые при исследовании.
Синдром Сетре Хотцена (Saethre-Chotzen) – редкое заболевание встречабщееся примерно в 1 на 25000-50000 новорожденных. Наследуется по аутосомно-доминантному типу. Наиболее общие признаки синдрома, наблюдаемые у трети пациентов – коронарный синостоз – брахицефалия, лицевая асимметрия, гипертелоризм, низкая линия роста волос, клинодактилия. В других, менее распространённых вариантах может быть плагиоцефалия, тригоноцефалия, позднее закрытие больших родничков и теменных отверстий, расширение турецкого седла, уплощение лобно-носового угла, супраглабеллярная депрессия, платибазия. Нос часто клювовидной формы, характерно отклонение носовой перегородки, высокое, арочное небо, часто неправильно сформированный прикус по причине сочетанных различных стоматологических аномалий. Возможен птоз, косоглазие, мелкие орбиты, телекантальные и эпикантальные складки, блефарофимоз, дакриостеноз, атрофия зрительного нерва, а так же гипотелоризм или гипертелоризм. Аномальная форма ушных раковин – они маленькие и низко расположены, снижен слух. Интеллект у таких пациентов часто в пределах нормы, в редких случаях наблюдается его снижение. Иногда встречается кожная синдактилия, может быть брахидактилия и клинодактилия. Характерно наличие широких больших пальцев с вальгусной деформацией. Кроме того может иметь место низкорослость, синостоз лучелоктевого сустава, короткие ключицы, малые подвздошные кости, крипторхизм и врожденные пороки сердца.